![]() The last day of February is Rare Disease Day, when we recognize the patients, researchers, and clinicians who deal with conditions that, despite their name, affect tens of millions of people around the world. Many rare diseases have a genetic basis and the rapid expansion of genetic testing by next-generation sequencing has transformed the field. Even with these powerful tools, we fail to diagnose more than half of patients with a suspected Mendelian disorder. Those patients and families endure a diagnostic odyssey — years of costly and sometimes invasive diagnostic testing. It’s still a challenging field. At my institution, we’ve been studying the genomic basis of rare disease for nearly a decade. We continue to make progress each and every year, but 2025 was a true outlier, a banner year for gene discovery in Mendelian disorders.
The last day of February is Rare Disease Day, when we recognize the patients, researchers, and clinicians who deal with conditions that, despite their name, affect tens of millions of people around the world. Many rare diseases have a genetic basis and the rapid expansion of genetic testing by next-generation sequencing has transformed the field. Even with these powerful tools, we fail to diagnose more than half of patients with a suspected Mendelian disorder. Those patients and families endure a diagnostic odyssey — years of costly and sometimes invasive diagnostic testing. It’s still a challenging field. At my institution, we’ve been studying the genomic basis of rare disease for nearly a decade. We continue to make progress each and every year, but 2025 was a true outlier, a banner year for gene discovery in Mendelian disorders.
Years-Long GeneMatcher Collaborations
First, several large and long-running international collaborations to establish novel disease-gene associations came to fruition at last. One of the more rewarding projects was EIF3A/EIF3B, genes in which heterozygous variants cause a neurodevelopmental disorder with craniofacial and cardiovascular features.
An Undiagnosed Noonan-like Syndrome: EIF3A/3B
Our patient was referred to research back in 2022. At the time, he was a 10-year-old boy with congenital heart disease, developmental delays, and dysmorphic features… essentially, a Noonan or RASopathy-like disorder which remained undiagnosed after a lot of testing, including trio exome sequencing. We performed trio genome sequencing and identified a promising lead, a de novo nonsense variant in EIF3B. This gene encodes the RNA-binding component of the eukaryotic translation initiation factor 3 (eIF-3) complex, which is required for several steps in the initiation of protein synthesis. That certainly seemed like a pathway in which mutations could cause a syndromic condition, though EIF3B was not associated with disease at that time.
We went to GeneMatcher and quickly connected with dozens of investigators who had similar patients. Clearly this was the answer for our patient and many others. Another institution already had a cohort in progress and was planning functional studies in zebrafish. We notified our family and expected that this work would be out somewhat soon, given the large number of patients and the importance of this gene. But it seemed to take forever. The project leader moved to a different institution. For a long time, it seemed like the project might be dead in the water, and we considered withdrawing to pursue it on our own. Then it found momentum again — there was a presentation at ACMG in 2024, and the full paper was accepted in September 2025… around 3 years after we joined. Granted, OMIM has yet to recognize the disease association for EIF3A or EIF3B, so these patients are likely still being missed by genetic testing.
A New Disease of Synaptic Dysfunction: UNC13A
Believe it or not, that wasn’t the longest-running collaboration that reached the finish line last year. Back in 2020, we took on another rare disease patient, a 9-year-old girl with weakness, delays, and epilepsy who had undergone extensive nondiagnostic testing dating back to 2011. We did trio genome sequencing, which uncovered compound-heterozygous variants in the UNC13A gene which is involved in neurotransmitter release at synaptic junctions. A few case reports had been published in 2016-2017 describing patients with variants in this gene in the setting of neurodevelopmental disease. That seemed promising, given our proband’s severe neurological issues. We went to GeneMatcher and got more than 50 matches. That’s often a good sign. One of those was a friend, Fowzan Alkuraya, who told me that there was already a collaboration in place, our patient was a pretty good fit, and they were still accepting new patients. So we joined the study, which was led by a group in Switzerland. This was in autumn of 2020.
There were functional studies to carry out and clinical features to compile. This might have felt less urgent because UNC13A was already recognized as a disease gene. We informed our family and did some necessary work, like segregation testing. The collaboration went along, at times slowly (I have notes from 2021 saying that we were in the cohort but I hadn’t heard from the study leaders in some time). In November of last year — five years after we joined the cohort — it was published in Nature Genetics. The paper describes 50 UNC13A patients — most biallelic, some monoalleic — which fall into three disease subtypes. There were 150 co-authors from 110 institutions! It’s a high-profile paper that truly establishes the disease phenotypic/genotypic spectrum.
Other Wins for GeneMatcher and Data Sharing
We contributed to a number of patient sharing, GeneMatcher, or other international matchmaking studies that were published last year:
- DDX17, an RNA helicase in which de novo mutations cause a neurodevelopmental disorder in humans and, wait for it, tadpoles (Brain 2025).
- SPOUT1, an RNA methyltransferase in which biallelic variants cause neurodevelopmental delays, a great effort led by my ASHG DLC buddy Avinash Dharmadhikari.
- ARF3, in which newly identified variants strengthen the link between Golgi fragmentation and brain malformations.
- SEPTIN2, which maintains the axon initial segment in neurogenesis, was linked to (autosomal recessive) cognitive impairment (Brain 2025)
- AUTS2, in which truncating mutations were known to cause neurodevelopmental disease, has a mutation hotspot in the EP300-binding region where missense variants cause disease, as nicely elucidated by my friend Kim Aldinger.
Almost all of these were projects that lasted 1-2 years if not longer, and it was rewarding to see them in print.
RNA-seq Provides Critical Answers in Rare Disease
We continue to apply new technologies and new analytical strategies to end the diagnostic odyssey for unsolved patients. RNA sequencing in particular has proven a useful tool for molecular diagnosis. We identified a splice-region variant that causes an atypical presentation of GNAS inactivation disorder. Pathogenic variants in this gene cause several syndromes depending on the variant type and location in the gene, whether it causes loss or gain of function, and whether it is germline or somatic in nature. Further complicating things is the fact that this locus is imprinted, so there’s a parent-of-origin effect. The family we studied had been followed here for more than a decade — first the mother, and then her daughter, were evaluated for unusual clinical features including a Madeling deformity (a wrist malformation). Clinical testing had identified a VUS in the GNAS gene shared by mother and daughter. It was a +5 splice region variant — not the essential splice site but often still critical for correct splicing. As we found with RNAseq, this variant had multiple effects.
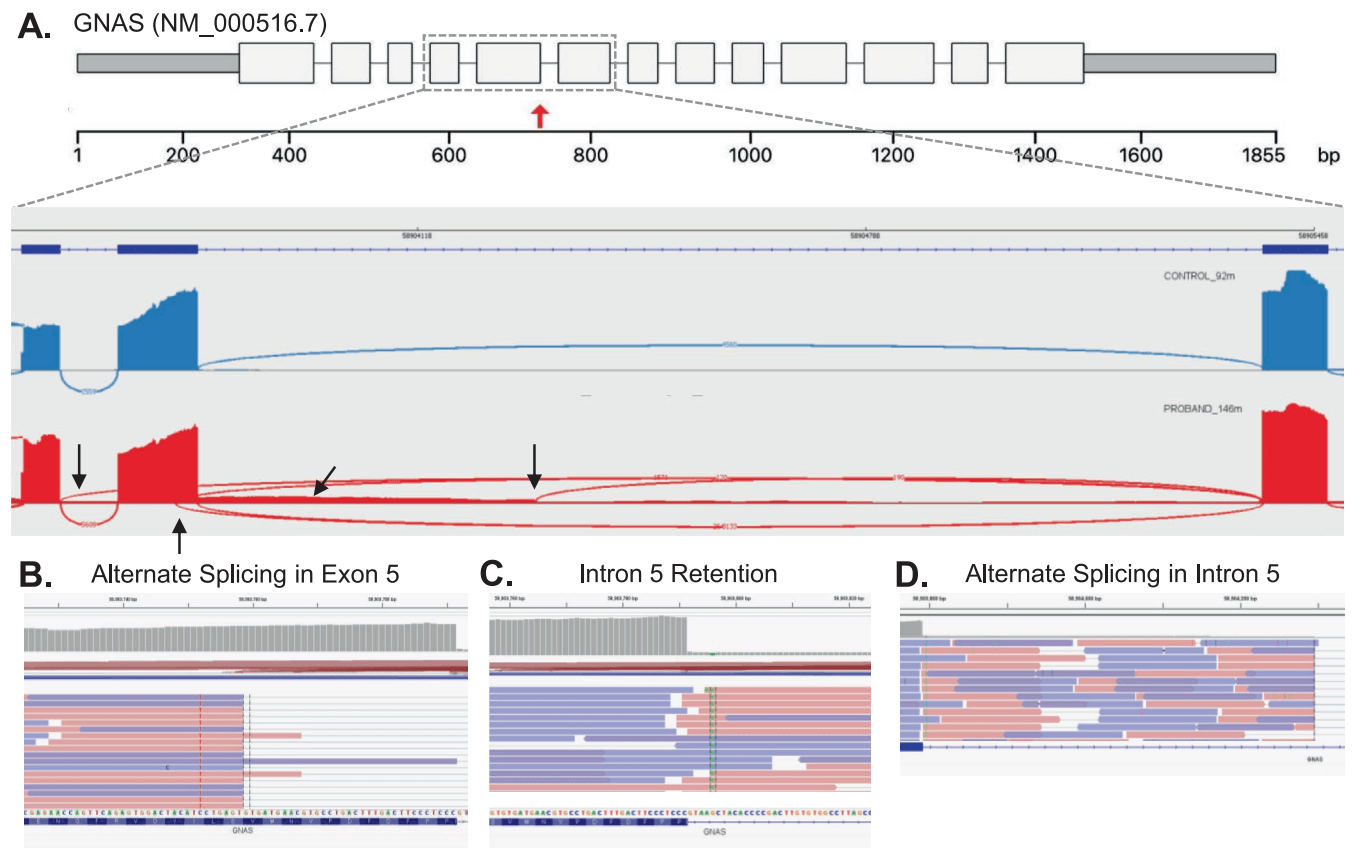
We had further work to do as well — for example, testing the mother’s parents which established that this variant was a de novo mutation in her, transmitted to her daughter. All of it came together in a nice case report at the American Journal of Medical Genetics Part A.
Like Father, Like Son
In a separate study, we enrolled a father and son who had a similar neuromuscular condition and shared a VUS in the splice region of ACTA1 (c.809-10C>A). This is outside the typical splice region area of influence, but the phenotype was a reasonable fit. However, splice prediction tools didn’t agree on the most likely consequence of this variant: one said it was a different splice site, and one said that splicing would be lost entirely. We wanted to determine which was the case, but needed access to the right tissue, muscle tissue. The proband had a recent muscle biopsy. The father had gotten one as well… thirty years ago. Our collaborators in the Center for Gene Therapy (Alayne Meyer and Kevin Flanigan) tracked down the muscle tissue in a freezer somewhere. We performed RNA-sequencing, and it worked, even for the father. Even better, it proved the effect of the variant:

Notice the incredibly high depth values reflecting the expression of this gene in muscle tissue. The effect of intron retention (i.e. loss of splicing) which was only apparent in about 20% of transcripts. Even so, it established the pathogenicity of the variant and consistency of the effect across thirty years of time.
The Near-Future of Rare Disease Genomics
We and our peer in the field of rare disease genomics are always pushing the boundaries. The technologies get better and better; so do the analysis tools and resources that we have at our disposal. As some of my examples show, RNA sequencing can be incredibly useful to both identify and determine the consequences of diagnostic variants. Currently, however, it’s mainly a research tool, not a diagnostic test. I’m part of the ClinGen Splicing Working Group, which has 20-30 members, and many of their institutions are working hard to validate RNA-seq for clinical use. That will be incredibly useful to help resolve some challenging cases.
Long-read sequencing and optical mapping technologies are also a key frontier in rare disease diagnostics. The cost of PacBio long-read sequencing continues to improve, and other platforms like Illumina Constellation have a lot of promise. This is all on the diagnostic side and does not even touch on the progress being made for therapeutics, especially gene therapy for rare diseases.
The bottom line is that 2025 was a great year for rare disease research, and 2026 might be even better.
Leave a Reply